Achievements of Members
Ram Babu1, Shruti Sinha2, Rutuja Kadam3, Manoj Sareen4, Shweta Juneja5
1Senior consultant, Medicine 2DNB Trainee, Family Medicine 3DNB Trainee Family Medicine 4Senior Consultant and Head of Department, Pathology 5Senior Consultant, Pathology, Jaipur Golden Hospital, Rohini, New Delhi.
Abstract- Kikuchi Fujimoto disease, also called as Histiocytic Necrotising lymphadenitis, is a rare, idiopathic self-limiting cause of lymphadenitis. It is most commonly seen in adult Asian females, younger than 40 years of age. The most common clinical manifestations of Kikuchi disease is cervical lymphadenopathy, with or without systemic signs and symptoms. We report a case of a 26 year old female who presented with mildly tender cervical lymphadenopathy which on excision biopsy revealed Necrotising Granulomatous Inflammation.
Introduction- The disease was first described in the year 1972, in Japan by Kikuchi and later in the same year by Fujimoto and colleagues independently. Also known as Histiocyctic Necrotising Lymphadenitis, it is a rare, self-limiting disorder, typically affecting the cervical group of lymph nodes. Cases have been reported mainly from Japan with few cases from Europe and Asia, with a female preponderance. It affects mostly young adults of less than 30 years of age.1-5
Case Report- A 26 year old female, known case of hypothyroidism, presented with multiple mildly tender cervical lymphadenopathy and malaise along with throat pain for 2 days. There was no history of fever/cough/night sweats/weight loss. Her vital parameters were within normal limits. CBC, LFT, KFT and Chest X Ray did not show any abnormality except for raised ESR (35). Patient was first started on antibiotics which gave no effective results. FNAC was done of the lymph nodes which was reported as reactive lymphadenitis favouring a diagnosis of tubercular lymphadenitis. Patient was started on Antitubercular treatment which was stopped in 3 days as the patient reported increase in the size of the lymph nodes. Patient was then taken up for excision biopsy, which showed expansion of mantle zone with folliculolysis, foci of geographic necrosis seen with sheets of signet ring shaped histiocytes. No immunohistochemistry was done as no atypical lymphocytes were seen. A diagnosis of Kikuchi- Fujimoto’s disease was offered. The biopsy material was sent for PCR which was negative for Mycobacterium Tuberculosis. Monospot test was negative (to rule out Infectious Mononucleosis), HIV, ANA profile, Anti ds DNA were found negative. The postoperative period was uneventful, and the patient was followed up for next 3 months. She was treated conservatively with NSAIDs, antiemetics as per need. She was given corticosteroids, which is recommended but efficacy is uncertain. Patient recovered completely and was symptom free in the next 3 months.
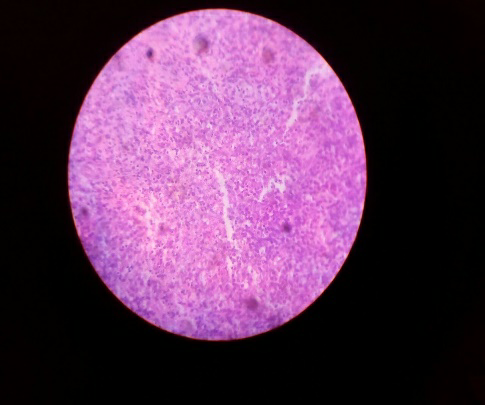
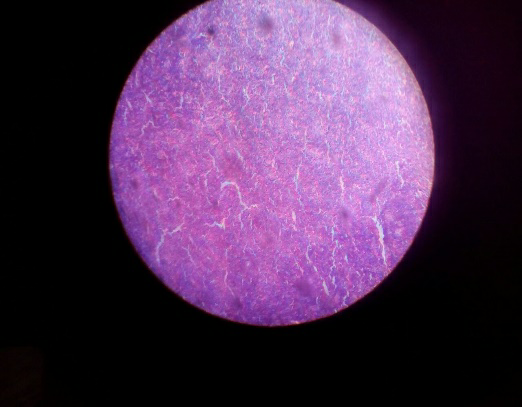
Fig1 &2: Cervical lymph node excision biopsy histopathological sections shows a fragmented lymph node showing expansion of mantle zone with folliculolysis. Foci of geographic necrosis is seen with sheets of signet shaped histiocytes.
Discussion- Kikuchi- Fujimoto disease is an enigmatic, benign and self-limiting condition characterized by regional tender lymphadenopathy, predominantly including the cervical region, with or without fever and night sweats.4 Lymphadenopathy may take several weeks to 6 months to resolve.
Described independently in 1972 by Kikuchi and Fujimoto et al, from Japan, till date most of the cases have been reported from East Asia, mostly affecting the younger population (20-30yrs) with a female preponderance.1-5 Of late few cases have been reported from the western countries.
The disease although is self-limiting, it can recur in about 3% of cases. Three deaths have been reported that occurred during the acute phase of generalized Kikuchi disease. One patient died from cardiac failure, second from effects of hepatic and pulmonary involvement, and the last from an acute lupus like syndrome. Another fatality has been reported in which Kikuchi disease appeared concurrently with SLE and was complicated with hemophagocytic syndrome and severe infection.
The cause of Kikuchi disease is unknown, although infectious and autoimmune etiologies have been proposed.7 The most favoured theory proposes that Kikuchi disease results when one or more unidentified agents trigger a self-limited autoimmune process. Lymphadenitis results from apoptotic cell death induced by cytotoxic T lymphocytes. Some Human Leukocyte antigen (HLA) class II genes are more frequent in patients of Kikuchi disease, suggesting a genetic predisposition to the proposed autoimmune response. Features that support a role for an infectious agent include the generally self-limited course of the disease and its frequent association with symptoms similar to those of upper respiratory tract infection. Several viral candidates have been proposed, including, Cytomegalovirus, Epstein-Barr virus, Human herpes virus, Varicella-Zoster virus, Parainfluenza virus, Parvovirus B19 and Paramyxovirus.8 However, serologic and molecular studies have failed to link Kikuchi disease to a specific pathogen, and more than one pathogen may be capable of triggering the characteristic hyperimmune reaction leading to Kikuchi disease. The differential diagnosis of cervical lymphadenitis is huge but the principal conditions to be distinguished are Lymphoma, Metastatic tumour from local or distant site, drainage from infective lesions in dependent skin and systemic conditions such as Infectious Mononucleosis, HIV, and most commonly Tuberculosis. There are several reports suggesting an association between Kikuchi’s disease and SLE.5 However, no convincing evidence is available to confirm such association.
The definitive diagnosis can be made on lymph node biopsy. Characteristic histopathological findings of KFD includes irregular central or paracortical areas of necrosis with abundant karyorrhectic debris. The nodal architecture can be distorted. Abundant histiocytes at the margin of the necrotic areas are seen. The karyorrhectic foci are formed by predominantly histiocytes and plasmacytoid monocytes but also immunoblasts and small and large lymphocytes. Plasma cells are either scarce or absent.3 Importantly, atypia in the reactive immunoblastic component is not uncommon and can be mistaken for lymphoma. The typical immunophenotype consists predominantly of T cells, with few B cells. Histiocyte associated antigens like CD68, lysozyme, myeloperoxidase are expressed by the histiocytes.3,4 Absence of significant number of plasma cells and/or hematoxylin bodies helps to distinguish Kikuchi disease from SLE.
Treatment of Kikuchi disease is generally supportive. NSAIDs may be used to alleviate lymph node tenderness and fever. The use of corticosteroids, such as prednisolone, has been recommended in several extranodal or generalized Kikuchi disease, and in cases of prolonged fever and annoying symptoms lasting more than 2 weeks despite NSAID therapy, as well as for recurrent disease. Indications for corticosteroids use include neurological involvement, hepatic involvement and severe lupus like syndrome.
Conclusion- Kikuchi Fujimoto’s disease is an uncommon, perhaps underdiagnosed condition of unknown etiology and excellent prognosis. Recognition of this condition is crucial as it may mimic tuberculous lymphadenitis, lymphoma, metastatic disease, or a local inflammatory/infective process. Awareness, not only by the clinicians but also the pathologists, may help prevent the misdiagnosis and overtreatment.
References
- Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytes: a clinicopathological study. Nippon Ketsueki Gakkai Zasshi. 1972; 35:379-80.
- Fujimoto Y. Cervical subacute necrotizing lymphadenitis. Naika. 1972; 30:920-7.
- Ioachim HL, Medeiros LJ, editors. Ioachim’s lymph node pathology. Lippincott Williams & Wilkins; 2009; 199-202.
- Hutchinson CB, Wang E. Kikuchi-fujimoto disease. Archives of pathology & laboratory medicine. 2010 Feb; 134(2):289-93.
- Bosch X, Guilabert A, Miquel R, Campo E. Enigmatic Kikuchi-Fujimoto disease: a comprehensive review. American Journal of Clinical Pathology. 2004 Jul 1; 122(1):141-52.
- Kampitak T. Fatal Kikuchi–Fujimoto disease associated with SLE and hemophagocytic syndrome: a case report. Clinical rheumatology. 2008 Aug 1; 27(8):1073.
- Sopeña B, Rivera A, Vázquez-Triñanes C, Fluiters E, González-Carreró J, del Pozo M, Freire M, Martínez-Vázquez C. Autoimmune manifestations of Kikuchi disease. InSeminars in arthritis and rheumatism 2012 Jun 30 (Vol. 41, No. 6, pp. 900-906). WB Saunders.
- Hudnall SD. Kikuchi-Fujimoto disease: is epstein-barr virus the culprit? 2000 June; 761-764.
- Singh YP, Agarwal V, Krishnani N, Misra R. Enthesitis-related arthritis in Kikuchi–Fujimoto disease. Modern rheumatology. 2008 Oct 1; 18(5):492-5.